

-
Vishal Gohil
- Professor, Biochemistry and Biophysics
- Focus Area: mitochondria, mitochondrial respiratory chain, metals, membranes, metabolism, mitochondrial disorders, copper, cardiolipin, elesclomol, menkes disease
- Office:
- ILSB 2146A
- Email:
- [email protected]
- Phone:
- 979-847-6138
Education
- Undergraduate Education
- B.Sc. 1995
- Graduate Education
- M.Sc. 1997
- Ph.D. 2005
- Postdoc. 2009
Areas of Expertise
- Cardiolipin
- Copper
- Elesclomol
- Menkes disease
- Metabolism
- Metals
- Membranes
- Mitochondria
- Mitochondrial disorders
- Mitochondrial respiratory chain
Professional Summary
My lab investigates the biochemical and genetic basis of mitochondrial dysfunctions in rare genetic disorders. This work is motivated by the observations that the molecular diagnosis of mitochondrial dysfunction in many genetic diseases is lacking, and no treatment is currently available for these debilitating and often fatal pediatric disorders. We apply the tools of genetics, genomics, and biochemistry in yeast, zebrafish, and mouse models to discover and characterize novel mitochondrial disease-causing genes. We then use our genetic models to perform targeted drug screens to develop therapeutic approaches for these debilitating human disorders.
Therapeutic Approaches for Mitochondrial Disorders
To determine the biochemical functions of our newly discovered mitochondrial disease genes, we construct yeast, zebrafish, and human cellular models by using gene-editing technologies. By characterizing these models, we have elucidated the role of copper in mitochondrial respiratory chain (MRC) biogenesis, organismal development, and human disease pathogenesis. We also use our genetic models to screen for therapeutic lead molecules and have discovered elesclomol, a copper ionophore, as a promising drug candidate for the treatment of disorders of copper metabolism such as the Menkes disease (See Figure 1). Inspired by our studies, elesclomol is currently being tested on children with Menkes disease and the initial results are very promising.
Guthrie, L.M., Soma, S., Yuan, S., Silva, A., Zulkifli, M., Snavely, T.C., Greene, H.F., Nunez, E., Lynch, B., De Ville, C., Shanbhag, V., Lopez, F.R., Acharya, A., Petris, M.J., Kim, B.E., Gohil, V.M. *, Sacchettini, J.C*. Elesclomol alleviates Menkes pathology and mortality by escorting Cu to cuproenzymes in mice. Science. 368:620-625 (2020).
Soma, S., Latimer, A.J., Chun, H., Vicary, A.C., Timbalia, S.A., Boulet, A., Rahn, J.J., Chan, S.S.L., Leary, S.C., Kim, B.E., Gitlin, J.D., Gohil, V.M. Elesclomol restores mitochondrial function in genetic models of copper deficiency. PNAS. 115:8161-8166 (2018).
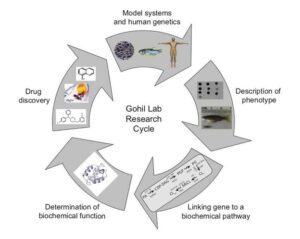
Figure 1: Experimental pipeline in the Gohil lab – from gene to drug discovery. See Soma et al., PNAS (2018) and Guthrie et al., Science (2020).
Mitochondrial respiratory chain biogenesis
Mitochondrial respiratory chain (MRC) is the primary means by which most eukaryotic organisms generate biological energy. Building the MRC is a complex process that requires hundreds of genes, many of which remain uncharacterized. We use an integrative approach based on clues from evolutionary history, human genetics, and nutrient-sensitized screens to systematically discover the “missing” factors in MRC biogenesis. We assign functions to these newly discovered genes by using biochemical, cellular, and molecular techniques. Using this approach, we have discovered proteins required for the delivery of copper to one of the MRC complex called cytochrome c oxidase (See Figure 2).
Garza NM, Griffin AT, Zulkifli M, Qiu C, Kaplan CD, Gohil VM. (2021) A genome wide copper-sensitized screen identifies novel regulators of mitochondrial cytochrome c oxidase activity. Journal of Biological Chemistry. 296:100485
Soma, S., Morgada, M.N., Naik, M.T., Boulet, A., Roesler, A.A., Dziuba,N., Ghosh, A., Yu, Q., Lindahl, P.A., Ames, J.B., Leary, S.C., Vila, A.J., Gohil, V.M. COA6 is structurally tuned to function as a thiol-disulfide oxidoreductase in copper delivery to mitochondrial cytochrome c oxidase. Cell Reports. 29:4114-4126.e5 (2019).
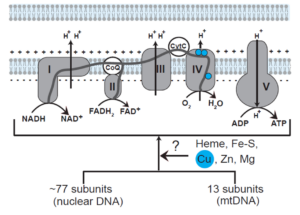
Figure 2: Missing factors in the MRC biogenesis pathway. See Garza et al., JBC (2021).
Phospholipid requirements for mitochondrial function and formation
The unique phospholipid milieu of the mitochondrial membrane can also influence the assembly and activity of the mitochondrial membrane protein complexes. We have constructed yeast mutants with defined perturbations in mitochondrial phospholipid composition to systematically determine the role of the three major mitochondrial phospholipids – phosphatidylcholine (PC), phosphatidylethanolamine (PE), and cardiolipin (CL) – in mitochondrial bioenergetics. Using these mutants, we demonstrated specific requirements of non-bilayer phospholipids, PE and CL, in MRC function. We have also used these mutants to uncover a specific requirement of CL for the human mitochondrial calcium and magnesium channels. These studies have uncovered the biochemical underpinnings of the pathology associated with Barth syndrome, a rare mitochondrial disorder of CL metabolism (See Figure 3).
Joshi A, Gohil VM. Cardiolipin deficiency leads to the destabilization of mitochondrial magnesium channel MRS2 in Barth syndrome. Human Molecular Genetics. 32:3353-3360 (2023).
Ghosh, S., Ball, W.B., Madaris, T.R., Srikantan, S., Madesh, M., Mootha, V.K., Gohil, V.M. An essential role for cardiolipin in the stability and function of the mitochondrial calcium uniporter. PNAS. 117:16383-16390 (2020).
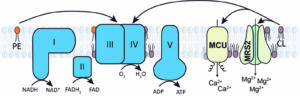
Figure 3: Phosphatidylethanolamine (PE) and cardiolipin (CL) are critical for the activity and stability of certain mitochondrial membrane proteins. See Ghosh et al., PNAS (2020) and Joshi et al., Hum. Mol. Genet. (2023).