

-
Sing Hoi Sze
- Associate Professor - Computer Science, Engineering, Biochemistry & Biophysics
- Focus Area: bioinformatics, computational biology, sequence analysis, biological networks, high-throughput sequencing
- Office:
- HRBB 328B
- Email:
- shsze@cse.tamu.edu
- Phone:
- 979-458-3396
Professional Summary
My work focuses on the application of computational techniques to solve problems in biology. Current research projects cover several areas in computational biology, including sequence alignment, motif finding, identifying gene clusters within genomes, biological network analysis, and high-throughput sequence assembly. Direct collaborations are maintained with biologists to solve real-life biology problems.
Sequence alignment
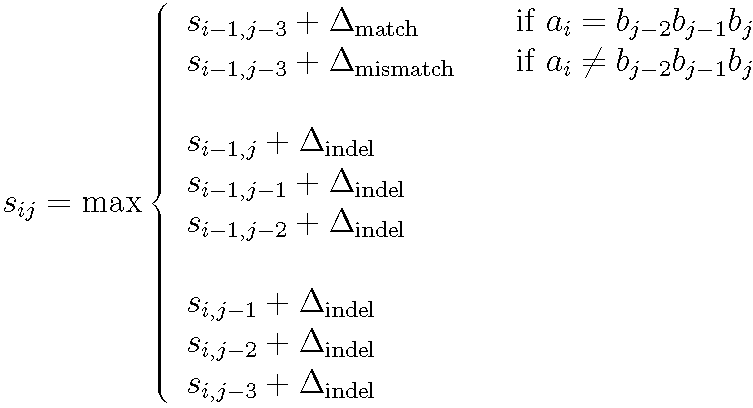
Recurrence for computing the optimal codon-based alignment score of a sequence A with i codons against a sequence B with j nucleotides. An exact match of three letters is assigned a positive score. A mismatch of at least one letter in a codon is assigned a negative score. An indel of either one, two or three letters is assigned a constant negative score.
We have proposed a polynomial time solvable formulation of multiple sequence alignment that achieves comparable performance with the best heuristics. This result challenges the common conception that the multiple alignment problem is hard and provides significant theoretical advances. A second project is based on the observation that alignment of divergent sequences can be improved by searching for and incorporating information in additional sequences from database search. A third project makes better use of horizontal information by taking into account alignment of neighboring residues when aligning two residues. The most recent project develops a codon-based alignment algorithm that identifies the frequency of RNA Polymerase II trigger loop variants from a set of mutant codons produced in deep sequencing of trigger loop amplicons.
Qiu C, Erinne OC, Dave JM, Cui P, Jin H, Muthukrishnan N, Tang LK, Babu SG, Lam KC, Vandeventer PJ, Strohner R, Van den Brulle J, Sze SH, Kaplan CD. High-Resolution Phenotypic Landscape of the RNA Polymerase II Trigger Loop. PLoS Genet. 2016 Nov 29;12(11):e1006321. doi: 10.1371/journal.pgen.1006321. eCollection 2016 Nov. PMID: 27898685
Motif finding
The focus is on developing new models and formulations to improve motif finding performance. We have developed an algorithm that integrates sample-driven and pattern-driven approaches to significantly improve computational time, an improved pattern-driven algorithm that achieves very good biological performance, an algorithm that finds DNA motifs by skipping nonconserved positions, and an algorithm to analyze expression of microRNAs from high-throughput sequencing data.
Zhu H, Hu F, Wang R, Zhou X, Sze SH, Liou LW, Barefoot A, Dickman M, Zhang X. Arabidopsis Argonaute10 specifically sequesters miR166/165 to regulate shoot apical meristem development. Cell. 2011 Apr 15;145(2):242-56. doi: 10.1016/j.cell.2011.03.024. PMID: 21496644
Identifying gene clusters within genomes
The focus is on investigating improved formulations to better model gene clusters within genomes. We have developed an algorithm that extracts statistically significant gene clusters from a genome with the help of gene ontology annotations, an algorithm that allows large-scale analysis of gene clustering in bacteria, and an algorithm that finds conserved gene clusters on multiple genomes.
Yang Q, Sze SH. Large-scale analysis of gene clustering in bacteria. Genome Res. 2008 Jun;18(6):949-56. doi: 10.1101/gr.072322.107. Epub 2008 Apr 4. PMID: 18390694
Biological network analysis
The focus is on developing efficient algorithms to extract functional substructures from biological networks and on identifying related subnetworks from a given network. We have investigated the problem of finding a path within a given graph that is most similar to another given path, with applications to identifying conserved pathways in different organisms. We have considered the problem of identifying complexes from a protein interaction network by considering different types of complexes separately. We have also considered the problem of finding conserved graphlets in protein interaction networks that correspond to network alignments.
Hsieh MF, Sze SH. Finding alignments of conserved graphlets in protein interaction networks. J Comput Biol. 2014 Mar;21(3):234-46. doi: 10.1089/cmb.2013.0130. Epub 2014 Feb 7. PMID: 24506222