

-
Directory Profile for Thomas Meek
- Professor, Biochemistry and Biophysics
- Focus Area: Determine chemical mechanisms of enzymes which are essential to infectious organisms, rational design of enzyme inhibitors
- Office:
- ILSB 2126
- Email:
- [email protected]
- Phone:
- 979-314-8286
Education
- Undergraduate Education
- B.S. in Chemistry, University of Virginia (1976)
- Graduate Education
- Ph.D. in Organic Chemistry, The Pennsylvania State University (1981)
- Postdoctoral Scholar, University of California, San Francisco (1981-1984)
Areas of Expertise
- Inhibitors
- Enzyme mechanisms
- Medicinal chemistry
- Infectious diseases
- Rational design
Professional Summary
Since 2014, the Meek lab’s mission is to elucidate the chemical mechanisms of enzymes that are validated drug targets and to utilize these mechanisms to develop rationally-designed inhibitors and inactivators of these enzymes. We research the following diseases: tuberculosis, African sleeping sickness, Chagas disease, malaria, and COVID-19 utilizing synthetic and medicinal chemistry, protein expression, crystallography, molecular modeling, mechanistic enzymology, and inhibitor characterization.
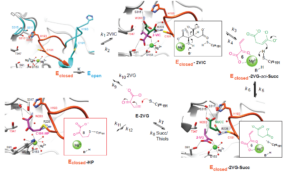
Crystal structure of (upper left) unliganded Mtb ICL1 (Eopen; blue) showing closure (Eclosed; orange) of the active-site loop upon substrate binding (k1). 2-VIC docked into ICL1 (Eclosed–2VIC), for which the inset shows the base-catalyzed, retro-aldol cleavage of 2-vinyl isocitrate (k3) forming aci-succinate (E–2VG–aci-Succ; green) and 2VG (6, purple). Protonation of aci-succinate by Cys191 forms succinate (k5) and Cys191 thiolate. Desorption of succinate (k7) provides steric freedom for Cys191-S– in Eclosed–2VG to proceed to either reaction with enzyme-bound 2VG (k11) (8, Eclosed-HP) or desorption (k9) of 2VG. In the k8 step, either succinate (succ) or DTT (thiols) may enter the active site.
Enzyme targets for tuberculosis
Isocitrate lyase (ICL) catalyzes the retro-aldol scission of D-isocitrate into glyoxylate and succinate, which initiates the glyoxylate shunt in mycobacteria. The glyoxylate shunt is an abridgment of the TCC cycle, providing mycobacteria a source of energy in a nutrient-poor environment. We have taken two approaches to develop inactivators of Mtb ICL. We have collaborated to obtain 2-vinyl-D-isocitrate (2-VIC), which we expected to be a mechanism-based inactivator. ICL-catalyzed scission of the C2-C3 bond produces vinyl-glyoxylate, a reactive Michael acceptor that forms a covalent bond with active-site Cys191 of ICL1.1 We have since discovered that the succinate analog cis-2,3-epoxysuccinate (EpS) is an exceptionally potent and selective inactivator of ICL1, which forms a covalent bond with Cys191 as confirmed by crystallography. EpS kills M. tuberculosis in cell culture under conditions in which the glyoxylate shunt activates. We are pursuing its analogs, which will have better cell penetration.
T. V. Pham, A. S. Murkin, M. M. Moynihan, L. Harris, P. C. Tyler, N. Shetty, J. C. Sacchettini, H.-I. Huang, T. D. Meek, Mechanism-based inactivator of isocitrate lyases 1 and 2 from Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 114, 7617–7622 (2017).
Parasitic diseases
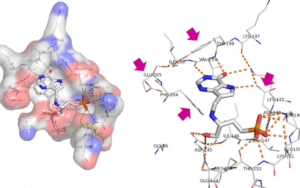
X-ray structure of T. cruzi HGXPRT bound to the purine-phospho-serinol transition-state analog (compound 53).
See ACS Biochem. 57, 3176–3190 (2018).
The parasitic protozoa Trypanosoma brucei and Trypanosoma cruzi are the respective causative agents of sleeping sickness and Chagas disease. These parasites cannot make purine nucleotides by biosynthesis de novo, unlike humans, and must rely on the salvage of purine bases from their hosts. A family of enzymes that are essential to the protozoa is the purine phosphoribosyltransferases (PPRTs), which catalyze the formation of XMP, GMP, and IMP from the corresponding base and PRPP. The trans-glycosylation reaction catalyzed by the PPRTs has a similar chemical mechanism to that of purine nucleoside phosphorylase (PNP), which has been a successful target for the development of highly potent, transition-state analog inhibitors (TSAIs). The discovery of these TSAIs was the result of the use of comprehensive kinetic isotope effects (KIEs) to structurally characterize the enzymatic transition, followed by the design and synthesis of compounds that mimic those structures. At least one of these TSAIs of PNP is a marketed drug. Analysis of the kinetic mechanisms and KIEs are underway for our prepared PPRTs. Some of the PNP TSAIs are exceptionally potent inhibitors of the PPRTs from all three species, and five are effective in a cellular model of Chagas disease.
X. Zhai and T. D. Meek, Catalytic mechanism of cruzain from Trypanosoma cruzi as determined from solvent kinetic isotope effects of steady-state and pre-steady-state kinetics. ACS Biochem. 57, 3176–3190 (2018).
Cysteine proteases of parasitic protozoa and coronavirus
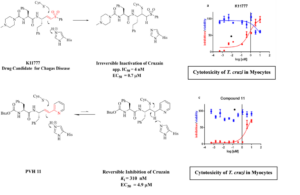
Irreversible and reversible inhibition of the cysteine protease cruzain from Trypanosoma cruzi. K11777 is a drug candidate for Chagas disease, shown with its effect in killing T. cruzi at sub-micromolar (700 nM) concentrations (red curves) in infected mouse myocytes (cell toxicity to murine cells, blue curves). Mechanism of reversible covalent inhibition by vinyl-pyridine (PVH 11), and its anti-T. cruzi efficacy in infected muring myocytes.
See JMC 63, 3298–3316 (2020).
Cruzain and falcipain are essential cysteine proteases of T. cruzi and P. falciparum, respectively. We have prepared both enzymes and completed an extensive kinetic analysis of their chemical mechanisms. For cruzain, we determined unusual, neutral protonation states of the active-site Cys and His residues, and we have elucidated all microscopic rate constraints of the double-displacement mechanism. These studies assisted in the design of two classes of inhibitors of unprecedented structures, peptidomimetic vinyl-heterocyclic (PVHIs) and masked aldehyde inhibitors (MAIs). Both classes of inhibitors are potent, reversible inhibitors of cruzain (some of falcipain), and many are highly effective in a cell model of Chagas disease. We are modifying the MAIs as pro-drugs to improve their pharmacokinetic properties, and we expect these pro-drugs to be suitable for animal model studies of Chagas disease.
B. C. Chenna, L. Li, D. M. Mellott, X. Zhai, J. L. Siqueira-Neto, C. Calvet Alvarez, J. A. Bernatchez, E. Desormeaux, E. Alvarez Hernandez, J. Gomez, J. H. McKerrow, J. Cruz-Reyes, T. D. Meek, Peptidomimetic vinyl heterocyclic inhibitors of cruzain effect antitrypanosomal activity. J. Med. Chem. 63, 3298–3316 (2020).
Cysteine protease 3CL-PR of SARS CoV-2
Potent, reversible, and selective inhibition of cysteine proteases is unprecedented, and these new classes of inhibitors are currently being adapted to inhibit an essential cysteine protease of the SARS-2 coronavirus, 3CL-PR, which we have expressed and purified in the Meek lab in May 2020. Through collaboration with the lab of Prof. Kent Tseng (UTMB), we have shown that four of our MAIs, which are effective in the cellular model of Chagas disease, kill the COVID-19 virus at low micromolar concentrations, as do other inhibitors of cruzain. This encourages the investigation of these new inhibitor classes vs. SARS CoV-2 3CL protease, which is now underway.
Publications
- View publications on PubMed